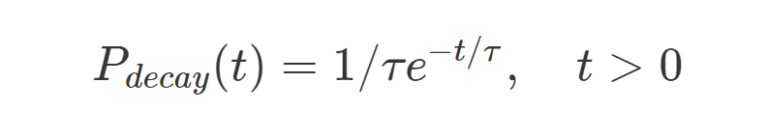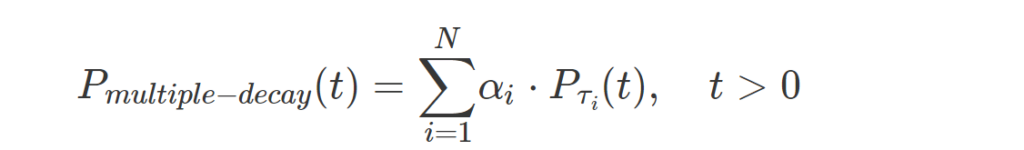Custom imaging products, sensors and software for low light level applications.
Fluorescence Lifetime Imaging Microscopy
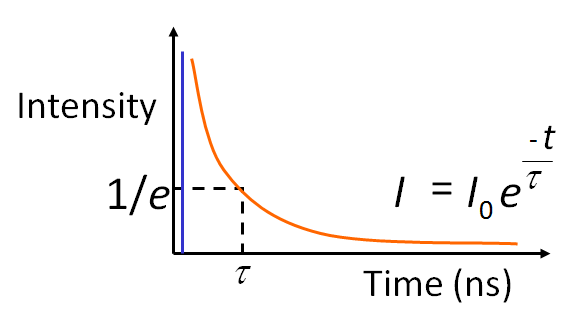
What is the Fluorescence lifetime? The fluorescence lifetime – the average decay time of a fluorescence molecule’s excited state – is a quantitative signature which can be used to probe structure and dynamics at micro- and nano scales. FLIM (Fluorescence Lifetime Imaging Microscopy) is used as a routine technique in cell biology to map the lifetime within living cells, tissues and whole organisms. The fluorescence lifetime is affected by a range of biophysical phenomena and hence the applications of FLIM are many: from ion imaging and oxygen imaging to studying cell function and cell disease in quantitative cell biology using FRET.
For fluorescent molecules the temporal decay can be assumed as an exponential decay probability function:
where t is time and τ is the excited state lifetime.
More complex fluorophores can be described using a multiple exponential probability density function:
where t is time, τi is the lifetime of each component and αi is the relative contribution of each component.
Why Measure Fluorescence Lifetime?
A key advantage of the fluorescence lifetime is that it is a basic physical parameter that does not change with variations in local fluorophore concentration and is independent of the fluorescence excitation. Hence the lifetime is a direct quantitative measure, and its measurement – in contrast to e.g. the recorded fluorescence intensity – does not require detailed calibrations. Excited state lifetimes are also independent of the optical path of the microscope, photobleaching (at least to first order), and the local fluorescence detection efficiency.
The fluorescence lifetime does change when the molecules undergo de-excitation through other processes than fluorescence such as dynamic quenching through molecular collisions with small soluble molecules like ions or oxygen (Stern-Volmer quenching) or energy transfer to a nearby molecule through FRET. As a result the fluorophores (in the excited state) lose their energy at a higher rate, causing a distinct decrease in the fluorescence lifetime. The measured rate of fluorescence is actually a summation of all of the rates of de-excitation. In this way the fluorescent lifetime mirrors any process in the micro-environment that quenches the fluorophores; and spatial differences in the amount of quenching reveals itself as contrast in a lifetime image.
Contact Us
5th floor, Leonard Springerlaan 19
9727KB Groningen
The Netherlands